Trustworthy prediction of super-resolution spatial gene expression#
Impute super-resolution spatial gene expression using GCN#
To predict pixel-level spatial gene expression \(\mathbf{Z}\in\mathbb{R}^{H_1\times W_1\times G}\), we train a deep neural network that maps the fused histology-gene embeddings \(\mathbf{F}=\mathrm{Concat}(\mathbf{U},\mathbf{V})\in\mathbb{R}^{H_1\times W_1\times (D+G_1)}\) to \(\mathbf{Z}\), using the spot-level count matrix \(\mathbf{Y}\) for weak supervision.
Our model is a graph convolutional network (GCN) with two shared graph-convolution layers (512 hidden units each). After the shared layers, each module (as indicated before) is predicted by its own head (a linear layer mapping 512 to the number of genes in the module). A dropout layer (p=0.5) is applied between the shared representation and each module-specific head to reduce overfitting.
To train and validate the model, and used the trained models to conduct prediction, run the following code:
python impute_final_split.py ${prefix} \
--cnts_train_name ${cnts_train_name}_seed_${seed}.csv \
--locs_train_name locs.csv \
--cnts_val_name ${cnts_val_name}_seed_${seed}.csv \
--locs_val_name locs.csv \
--epochs=400 --device='cuda' --n_states=5
- Input:
embeddings-combined-train.pickle: combined gene expression and histology features derived from training data.embeddings-combined-all.pickle: combined gene expression and histology features derived from original data.cnts_train_seed_1.csv: training set.cnts_val_seed_1.csv: validation set.locs.csv: spot location matrix paired with training and validation set (same for countsplit).gene-names.txt: genes selected for enhancement.radius.txt: rescaled spot radius.
- Parameters:
${prefix}: directory to the folder containing the image, i.e.data/.--cnts_train_name: file name of the training count data.--locs_train_name: file name of the spot location of the training data (must be paired with the training count data).--cnts_val_name: file name of the validation count data.--locs_val_name: file name of the spot location of the validation data (must be paired with the training count data).--epochs: number of epochs when training the model.--device: device used for training, validation and prediction. The use of GPU is strongly recommended.--n_states: number of states (number of independent models trained, validated and used for prediction).
Output:
Prediction_val/cnts-super/: directory containing enhancement results, with each gene saved asGENE.pickle.
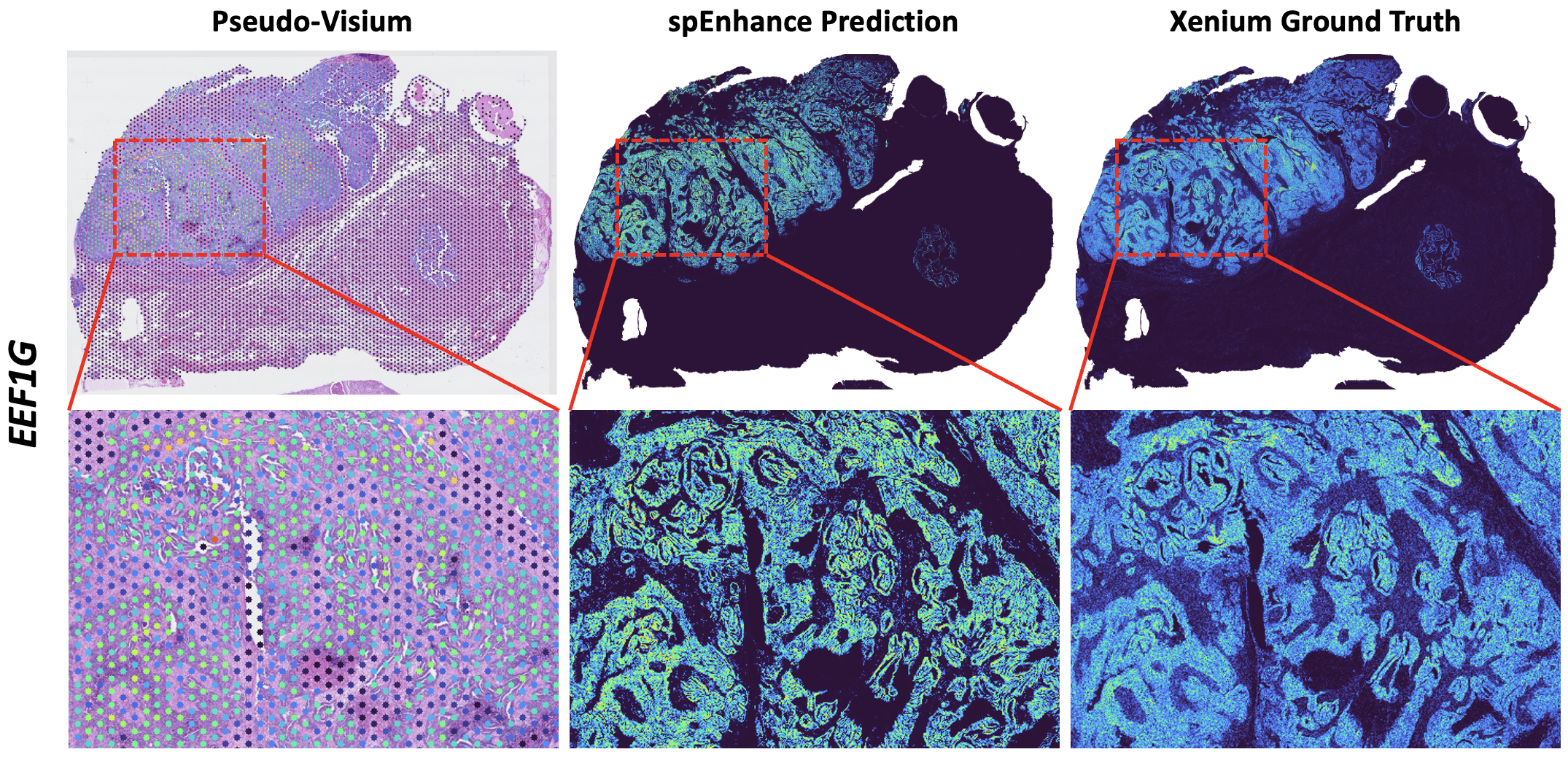
Error quantification to prevent model overfitting#
To avoid overfitting and ensure computational efficiency, the spEnhance count-split (spEnhance-CS) framework identifies the optimal training epoch – the point at which the validation loss reaches its minimum.
Given validation-level spot counts \(Y_{s,g}^{(\mathrm{val})}\) for spot \(s\) and gene \(g\), spEnhance-CS proceeds in two steps:
Impute super-resolution using validation embeddings. The model first uses embeddings learned from the validation data to generate imputed super-reolustion outputs and then aggreate into spot level, denoted by:
\[\widehat{\lambda}_{s,g}\]Qunatify error at both local and golbal levels.
Local error: We compute the Pearson predictive residual:
\[\widehat{S}_{s,g}=\sqrt{Y_{s,g}^{(\mathrm{val})}}-\sqrt{\widehat{\lambda}_{s,g}}\]Global error (gene level): Derived by comparing imputed values with validation data across spots using metrics including SSIM, RMSE, and PCC.
Run the following code to generate local error visualization and global error statistics.
python error_quant.py --prefix ${prefix} \
--cnts_val_name ${cnts_val_name}_seed_${seed}.csv \
--locs_val_name locs.csv
- Input:
embeddings-combined-val.pickle: combined gene expression and histology features derived from validation data.gene-names.txt: genes selected for enhancement.radius.txt: rescaled spot radius.cnts_val_seed_1.csv: validation set.locs.csv: spot location matrix paired with validation set.
- Parameters:
${prefix}: directory to the folder containing the image, i.e.data/.--cnts_val_name: file name of the validation count data.--locs_val_name: file name of the spot location of the validation data (must be paired with the training count data).
- Output:
local_uncertainty_plots/: directory containing local error quantification results, with each gene saved asGENE.png.df_spot_rank.csv: file containing global error for each gene.